
Sequencing techniques - let there be light!
Paulia Smaruj
Wiceprezes, Koło Naukowe Biologii Molekularnej UW
Pierwsze techniki sekwencjonowania były przełomowe. Pokazały człowiekowi, że poznanie dokładnej kolejności nukleotydów w makromolekułach DNA jest możliwe. Co było kolejnym celem? Robienie tego szybciej, z większą dokładnością i mniejszym nakładem finansowym. Proszę Państwa, wkraczamy w technologie NGS – Next Generation Sequencing, zwane też technikami wysokoprzepustowymi (High Throughput Sequencing Technologies).
Lakoniczna charakterystyka technik sekwencjonowania II generacji:
- Generowanie milionów krótkich odczytów równolegle.
- Zwiększenie szybkości sekwencjonowania w porównaniu z technologiami I generacji.
- Obniżenie kosztów sekwencjonowania.
- Wynik sekwencjonowania jest otrzymywany bez potrzeby wykonywania elektroforezy DNA.
„I stała się światłość” – pirosekwencjonowanie
Sekwencjonowanie Roche/454 pojawiło się na rynku w 2005 (choć sam pomysł powstał jeszcze w latach 90.). Technika pirosekwencjonowania oparta jest o detekcję pirofosforanu (PPi), który ulega wydzieleniu przy inkorporacji każdego nukleotydu nowosyntetyzowanej nici (czyli podobnie jak metoda dideoksy, zalicza się je do SBS – sequencing by synthesis).
Jednak zacznijmy od początku. Wyobraźmy sobie krótki fragment DNA (jeden z olbrzymiej liczby znajdujących się w bibliotece DNA, którą wcześniej uzyskaliśmy). Naszą makromolekułę przyłączamy do kulki mikroskopijnych rozmiarów (jest zbudowana z substancji paramagnetycznej, w literaturze angielskiej określa się ją jako koralik – bead). DNA jest przyłączone do kulki przez specyficzną sekwencją, określaną jako adapter. Następnym krokiem jest przeprowadzenie nietypowego rodzaju reakcji PCR – w emulsji woda-olej (ang. water-in-oil emulsion PCR, emPCR). Idea jest taka, żeby każdy koralik znalazł się w kropelce wody zwieszonej w oleju (proporcje są tak dobierane, by statystycznie w jednej kropli znalazła się jedna kulka, nie więcej). Przeprowadzamy PCR, skutkiem czego powierzchnia koralika jest pokryta zamplifikowanymi klonami wyjściowej sekwencji. Następnie pokrywamy mieszaniną płytkę zawierającą mnóstwo dołków (ang. picotiter plate) i znów podobna zasada: jedna kulka powinna trafić do jednego dołka.
Teraz przechodzimy do pirosekwencjonowania sensu stricto: po płytce rozprowadzamy mieszaninę enzymów i dNTP. Co dokładnie dzieje się w każdym z dołków? Zachodzą następujące reakcję:
- Inkorporacji dNTP komplementarnego do matrycy (zwróćmy uwagę na powstający nieorganiczny pirofosforan):
(DNA)n + dNTP → (DNA)n+1 + PPi + H+
- Uwolniony pirofosforan ulega przekształceniu do trifosforanu adenozyny (ATP), reakcję katalizuje sulfurylaza ATP:
PPi + APS → ATP + SO42–
- ATP jako kofaktor enzymu lucyferazy pozwala na zajście reakcji utlenienia lucyferyny do lucyferazy, której produktem jest impuls światła:
ATP + lucyferyna + O2 → AMP + PPi + oksylucyferyna + CO2 + ℎ?
Oznaczenia we wzorach: AMP – adenozynomonofosforan; SO42– – anion siarczanowy; AMP – monofosforan adenozyny; CO2 – tlenek węgla (IV); ℎ? – światło.
Detekcji podlega wydzielany impuls światła (technicznie robi to urządzenie określane jako charged couple device (CCD) znajdujące się pod dołkami). Wynikiem jest odczyt o długości 400-500 nt.
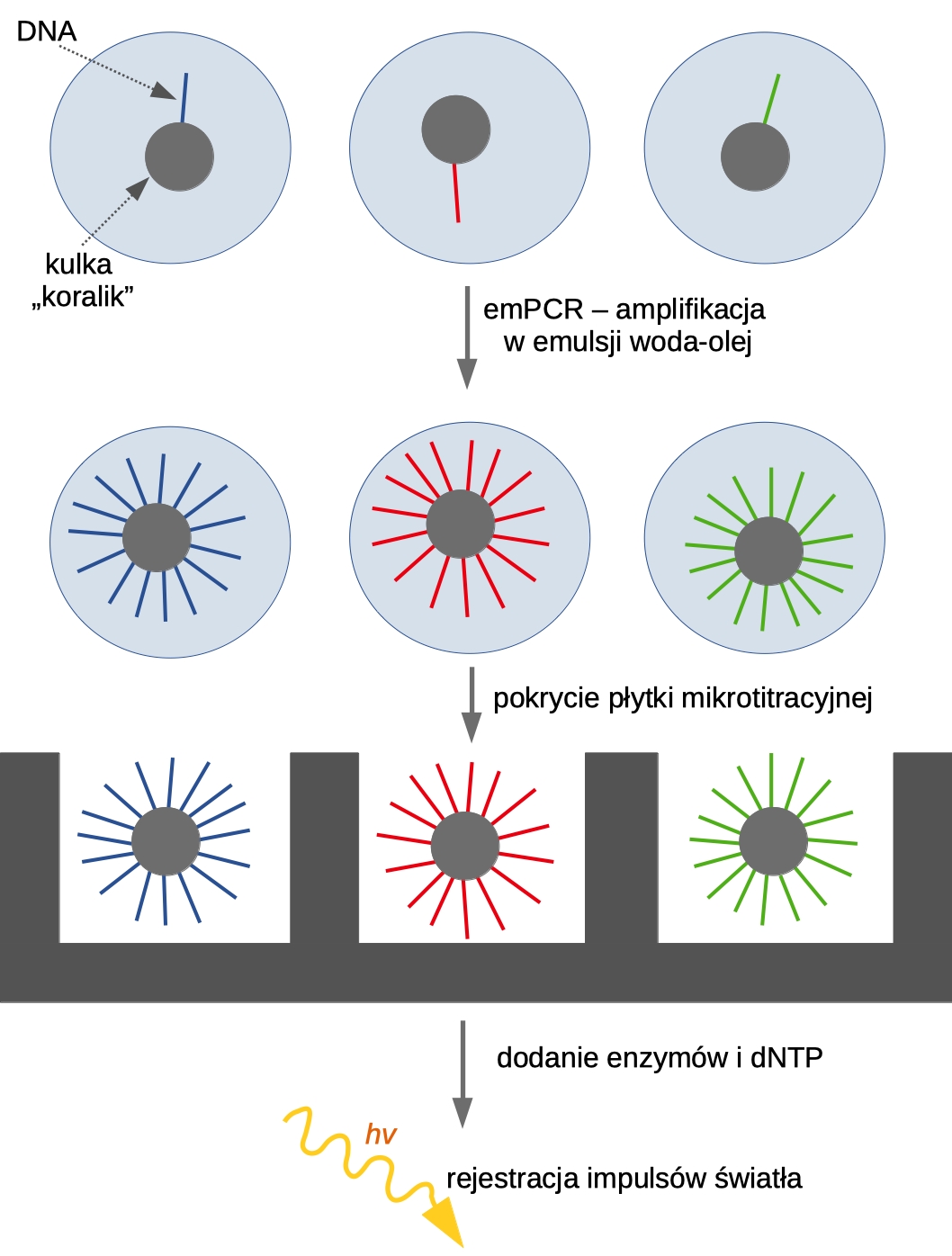
Ryc.1. Poszczególne etapy techniki Roche/454. Fragmenty DNA są początkowo unieruchamiane na mikroskopijnych kulkach, a następnie amplifikowane metodą emPCR. Każdy koralik zostaje w ten sposób pokryty klonami wyjściowej sekwencji. Mieszanina zostaje rozprowadzona po płytce w taki sposób, by jedna kulka trafiła do jednego dołka. Po zainicjowaniu reakcji polimeryzacji, impulsy świetlne są rejestrowane przez urządzenie zlokalizowane pod dołkami.
A teraz rozszerzmy perspektywę, wyjdźmy spoza dołka. Na płytce skewencjonatora znajdują się miliony takich samych dołków, w których zachodzą identyczne relacje chemiczne. Odczyt sekwencji fragmentów DNA pokrywających wszystkie kulki znajdujących się we wszystkich dołkach odbywa się równolegle i w czasie rzeczywistym. Czy już wyczuwacie jak duże możliwości daje ta technika?
Pirosekwencjonwanie ma jednak kilka wad, które po krótce wymienimy:
- Otrzymując tak krótkie odczyty musimy dysponować odpowiednimi możliwościami z zakresu bioinformatyki, które pomogą nam uzyskać dłuższe sekwencje (proces nazywany jest składaniem).
- Każdy nukleotyd wraz z enzymami jest dodawany osobno, co zwiększa koszty.
- Co w przypadku, gdy mamy dwa takie same nukleotydy z rzędu? Powiedzmy AA. Generalnie większa intensywność wydzielonego światła odpowiada odpowiada większej liczbie wstawionych nukleotydów. Czyli intensywność promieniowania, które zarejestruje detektor będzie 2 razy większa w przypadku AA niż przy pojedynczym A. Wyobraźmy sobie jednak, że w naszej sekwencji zdarzył się ciąg kilku-kilkunastu takich samych nukleotydów (więcej niż 5-6), co często określa się jako poli(N), gdzie N oznacza dowolną zasadę azotową. Niestety przy tak dużej liczbie takich samych nukleotydów przyrost intensywności wykrywanego promieniowania nie jest proporcjonalny do długości homopolimeru. Więc określenie, czy sekwencja zawierała 10 czy 11 adenin może okazać się niemożliwe przy pomocy tej techniki.
Technika pirosekwencjonowania była niesamowitym przełomem – otworzyła oczy i umysły na zupełnie nowe, dziesiątki razy szybsze strategie poznania sekwencji kwasów nukleinowych. W przenośni i dosłownie – stała się światłość…
Bibliografia:
- Harrington CT, Lin EI, Olson MT, Eshleman JR. 2013. Fundamentals of Pyrosequencing. Archives of Pathology & Laboratory Medicine, 137: 1296–1303.
- Heather JM, Chain B. 2016. The sequence of sequencers: The history of sequencing DNA. Genomics 107(1): 1–8.
- Kchouk M, Gibrat JF, Elloumi M. 2017. Generations of Sequencing Technologies: From First to Next Generation. Biology and Medicine 9: 395.